Nazwa projektu: ABCFEH
Lider projektu: Tamás Hegedűs
Instytucja macierzysta: MTA-SE Grupa Badawcza Biofizyki Molekularnej, Węgierska Akademia Nauk, Budapeszt, Węgry oraz Wydział Biofizyki i Biologii Radiologicznej, Uniwersytet Semmelweis, Budapeszt, Węgry.
Mukowiscydoza jest najbardziej powszechną chorobą monogenową w populacji kaukaskiej i charakteryzuje się wysoką śmiertelnością. Śmiertelność związana jest z niemożnością wypływu chlorków z komórki do płynu powierzchniowego dróg oddechowych, co prowadzi do powstania nadmiernie gęstego i lepkiego śluzu. Zjawisko to prowadzi do dysfunkcjonalnego bicia rzęsek i upośledza oczyszczanie z kurzu i bakterii. Podobne procesy powodują problemy w układzie pokarmowym (jelito, trzustka). Z drugiej strony, w skórze nie ma resorpcji chlorkowej z potu, więc pacjenci tracą ważny jon. Nadmierną utratę soli można łatwo wyczuć zmysłem smaku, a służyła ona do diagnozy, związanej z czarnoksięstwem pod koniec XV wieku i wyrażonej w irlandzkim przysłowiu: “Biada temu dziecku, które smakuje słono, gdy całuje się je w czoło.Rzucono na nie urok i wkrótce musi umrzeć”.

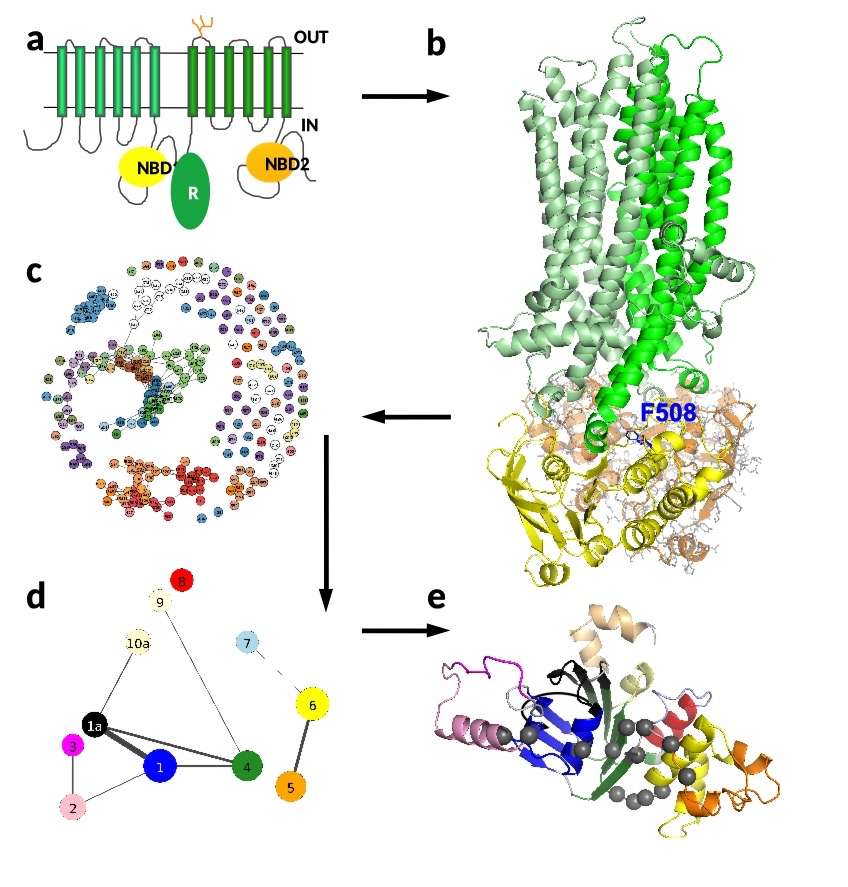
Mukowiscydoza jest spowodowana mutacjami w genie białka CFTR, które jest kanałem chlorkowym w błonie komórkowej. Kiedy to białko jest zmutowane, wpływa to na jego strukturę, zwijanie i dynamikę. Głównym celem w tej dziedzinie jest zaprojektowanie leków, które przywrócą normalne strukturalne i dynamiczne właściwości CFTR i dostarczą ten kanał do błony komórkowej. Znajomość struktury białek ułatwia projektowanie i ulepszanie leków (a). Struktura CFTR składa się z 6+6 helis transmembranowych przecinających błonę komórkową, dwóch wewnątrzkomórkowych domen wiążących nukleotydy/ATP (NBD1 i NBD2). Przedstawiono wszystkie atomy NBD2, ale pozostałe części zostały uproszczone w celu stworzenia wizualizacji (b). Najczęściej zmutowany aminokwas F508 jest zaznaczony niebieskimi liniami. Usunięcie tej fenyloalaniny (ΔF508) zakłóca nie tylko interakcję między helisami NBD1 (żółtymi) i transmembranowymi (zielonymi), ale także wpływa na stabilność NBD1.
Ponieważ eksperymentalna obserwacja ruchów atomów w białkach jest w większości przypadków niezwykle trudna lub wręcz niemożliwa, stosujemy metody in silico do obliczenia ruchów w oparciu o fizykę, w celu uzyskania filmu na poziomie molekularnym (http://www.hegelab.org/md.html). O ile łatwo jest obliczyć ruch jednego lub dwóch atomów, o tyle w przypadku setek tysięcy atomów w białku jest to już większe wyzwanie. Jest to spowodowane nie tylko dużą liczbą atomów, ale również skomplikowanymi, odległymi interakcjami pomiędzy atomami, takimi jak elektrostatyka. Dlatego tak zwane obliczenia dynamiki molekularnej wymagają wysokowydajnych zasobów obliczeniowych (np. komputerów z dużą liczbą procesorów i kilku wysokowydajnych procesorów graficznych).
Analiza stanowi również wyzwanie, opierając się na różnych metodach statystycznych i sieciowych. Badając charakter zbiorowisk w sieci aminokwasów (c, d) można zidentyfikować te krytyczne (e), które mogą umożliwić rozprzestrzenianie potencjalnie szkodliwych mutacji w całej, szerszej wspólnocie aminokwasów. Następnie stają się one potencjalnymi celami dla leków, które mogą powstrzymać to rozprzestrzenianie się.
W oparciu o nasze wyniki, wiązanie się tysięcy cząsteczek z tym krytycznym miejscem może zostać przetestowane in silico. Najlepsze trafienia, które zwykle obejmują tylko dziesiątki kandydatów na leki, mogą być dalej testowane przy użyciu metod eksperymentalnych. Eksperymentalnie skuteczne i nietoksyczne cząsteczki stają się wiodącymi cząsteczkami do dalszego rozwoju i badań klinicznych. Podsumowując, wspólna wiedza z zakresu biologii, fizyki i matematyki może pomóc w zrozumieniu wpływu mutacji na strukturę i dynamikę białek, w identyfikacji miejsca wiązania leków i potencjalnie w rozwoju terapii chorób, takich jak mukowiscydoza.